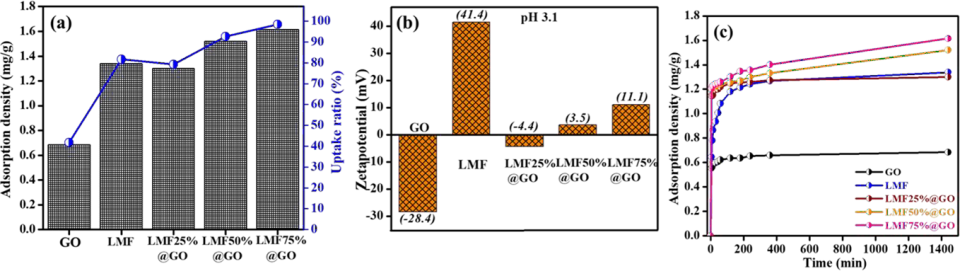RemPlex Summit Technical Sessions

General Technical Session
Nov. 12, 2021, 10:45 am - 12:15 pm
Organizer(s): Hilary Emerson and Nik Qafoku, Pacific Northwest National Laboratory, Richland, Washington
The General Session is open to all topics relevant to the remediation of complex sites. Examples include characterization studies, contaminant fate and transport, geochemical and microbiological contaminant controls over contaminant mobility, conceptual site model development, subsurface contaminant interactions, remedy development, and others. Presentations relevant to case studies are also encouraged.
Nov. 12 General Technical Session Recording Now Available |
|
|
Theme: Tc/I Fate in the presence of Mn-oxides
Speaker #1
Seonyi Namgung
Yonsei University, Seoul, Republic of Korea
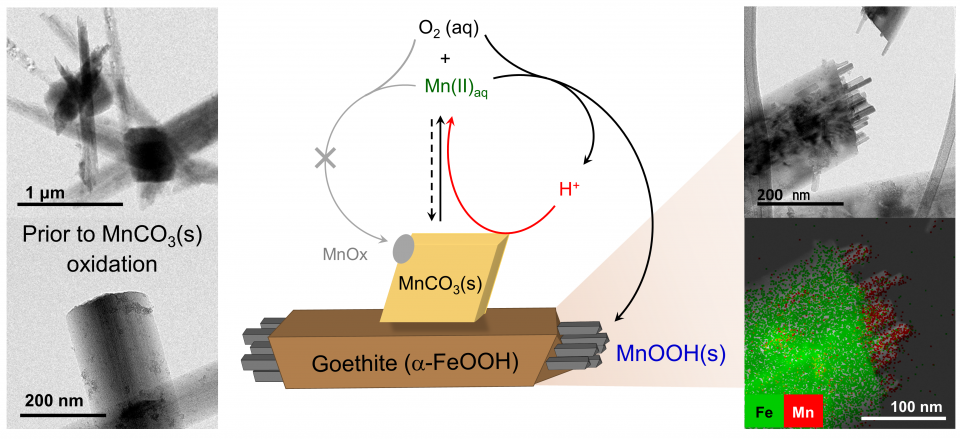
Effect of Goethite as a Solid Substrate on Oxidative Conversion of Rhodochrosite to Groutite-like Mn Oxides
Co-authors: Giehyeon Lee, Yonsei University
Abstract
Manganese (oxyhydr)oxides (referred to hereafter as oxides) are among the most powerful natural oxidants and scavengers, greatly responsible for the fate and transport of various nutrients, contaminants, and other substances in the environment. These oxides are commonly found as a variety of solid phases with unique physico-chemical properties and reactivities to the contaminants depending on their crystal structure, structural defects and Mn valence. Thereby, there have been extensive studies on geochemical and microbiological processes of diverse Mn oxides formation via the oxidation of dissolved Mn(II). However, the oxidative conversion of solid phase Mn(II) to Mn(III/IV) oxides is still poorly understood. Here, we examined the oxidation of rhodochrosite (MnCO3), one of the most common Mn(II) solid phases in nature, by dissolved oxygen and the subsequent formation of Mn oxides with a special focus on the geochemical roles of goethite as a model solid substrate ubiquitous in the environment. In contrast to the limited oxidation without goethite, the rhodochrosite oxidation was significantly accelerated in the presence of goethite and consequently induced heteroepitaxial growth of groutite (α-MnOOH)-like Mn oxides on the goethite (α -FeOOH) tip surfaces. Our results discussed the geochemical processes accelerating rhodochrosite oxidation with the critical roles of goethite substrate. This study provides new insights into the geochemical behaviors of redox-sensitive solid phases in complex subsurface and surface environments and thereby expands our understanding of the geochemical role of manganese on the fate and transport of various contaminants.
Speaker #2
Vasileios Anagnostopoulos
University of Central Florida, Orlando, Florida
Technetium Proliferation Under Anoxic Conditions by Manganese Oxides: the Case of Solid-solid Interactions
Co-authors: Jordan Stanberry and Ilana Szlamkowicz, University of Central Florida
Abstract
Manganese oxides are ubiquitous oxidizing agents in the environment that have been found to oxidize a wide variety of metal and metalloid contaminants in the natural environment. While manganese oxides are found at lower environmental concentrations than some of their iron counterparts, their control over the redox potential of the system often outweighs their concentration due to two factors: the high reduction potential of the Mn(IV or III)/Mn(II) redox couple and the redox cycling of manganese by microorganisms. The later factor ensures that high valent manganese concentrations remain relatively stable during redox interactions even under anoxic conditions.
Technetium-99 (T1/2 = 2.13*105 years) is a risk driving radionuclide for many Department of Energy Nuclear (DOE) sites including the Hanford site, Savannah River and Oak Ridge sites. Past improper handling of legacy waste has led to accidental or intentional the release of Tc-99 into the environment at these sites. Tc-99’s environmental mobility is governed by its oxidation state: Tc(IV)O4- predominates under oxidizing conditions and is highly mobile, while Tc(IV)O2 predominates under reducing conditions and is relatively immobile environmentally. Because of this remediation efforts have historically relied on the reductive immobilization of TcO4- to TcO2.
Despite the ubiquity of manganese oxides and their propensity to oxidize a wide variety of environmental contaminants, the oxidative dissolution of TcO2 by manganese oxides had not been previously studied. In this study, we investigated for the first time the redox reaction between these two insoluble minerals. We performed batch dissolution experiments in an anaerobic glovebox (<0.1 ppm O2), at multiple environmentally relevant pH values (pH 6.5 buffered and pH 8.0, in the presence and absence of HCO3-), using three different manganese oxides: birnessite (MnO2), manganite (MnOOH) and bixbyite (Mn2O3). We investigated the effect of manganese oxide to TcO2 ratios and the effect of sorption of divalent cations on the manganese minerals and their redox capacity.
Our results showed that all three minerals are capable of rapidly oxidizing the immobile TcO2 phase to the highly mobile TcO4- phase even under anoxic conditions. As direct electron transfer from one solid to another is unlikely, we hypothesized that short lived aqueous Tc(IV) intermediates are formed which sorb to the manganese oxides and are rapidly oxidized to Tc(VII) species which are immediately released in the aqueous phase. Furthermore, we observed an increased oxidation rate of TcO2 when HCO3- was present at pH 8 and the sorption of divalent cations significantly decreases the percent of TcO2 oxidized by the manganese oxides, most probably due to the sorption of these cations on the redox active centers of the minerals.
Overall, manganese oxides exert significant geochemical controls on contaminant behavior in the environment, and specifically Tc, hence their presence needs to be considered taken into account wen planning remediation strategies.
Speaker #3
Jordan Stanberry
University of Central Florida, Orlando, Florida
Manganese(III)-Ligand Complexes as Major Oxidizing Agents Affecting TcO2 Stability Under Anaerobic Conditions
Abstract
Technetium-99 (T1/2 = 2.13*105 years) is one of the main risk driving contaminants at several Department of Energy nuclear sites across the United States. Technetium-99 is a redox sensitive radionuclide whose environmental mobility is largely controlled by its oxidation state. Reduced forms of technetium, such as Tc(IV)O2, are relatively insoluble and environmentally immobile. On the other hand, the predominant technetium species under oxidizing conditions is Tc(VII)O4-, which does not sorb onto most mineral or soil surfaces, is highly soluble and environmentally mobile. Because of this, remediation of Tc-99 in the subsurface has relied historically on the reductive immobilization of TcO4- to TcO2.
The success of remediation of Tc-99 by reductive immobilization relies on the stability of TcO2 in subsurface environments where the method is employed. The presence of localized oxidizing agents could pose a problem for the long-term efficacy of the method. One such oxidizing agent is aqueous Mn(III)-ligand complexes. While free Mn(III) in aqueous solutions is not stable and will rapidly disproportionate to Mn2+ and solid MnO2, the presence of complexing ligands can stabilize in Mn(III) in the aqueous phase. Mn(III)-ligand complexes have been detected in suboxic and anoxic subsurface environments. Despite their prevalence in suboxic and anoxic zones and their high redox potential, the redox interactions of Mn(III)-ligand complexes with TcO¬2 have never been studied before.
We investigated the oxidative dissolution of TcO2 by Mn(III)-pyrophosphate under anaerobic conditions (anaerobic glovebox, <0.1 ppm O2) in batch experiments. The effects of pH, pyrophosphate to Mn(III) concentration ratio, and the total Mn(III) concentration on the oxidation of 200µg of TcO2 were quantified. All solutions were prior degassed using sonication-vacuum methods and all experiments were performed in triplicates. The concentration of Tc-99 released in the aqueous phase was monitored using Liquid Scintillation Counting, and the disappearance of Mn(III)-PP was monitored using UV-vis. Total Mn in the aqueous phase was measured using ICP-MS, and X-ray diffraction and Scanning Electron Microscopy were used to identify solid products formed in the redox reaction.
Our initial results indicate that as low as 0.6 mM Mn(III)-PP is capable of rapidly and substantially oxidizing TcO2 to the more mobile TcO4-, despite the absence of oxygen. In most of our experiments the redox reaction plateaued in approximately 3 hours. The rate and percentage of TcO2 oxidation decrease as the pyrophosphate:Mn(III) ratio increases. However, a minimum ratio of 4:1 is required for the stability of the complex. On the other hand, the effect of pH in the range of 6-8 on the kinetics of the reaction are negligible. The rate and extent of TcO2 oxidative dissolution increases approximately linearly with increasing initial Mn(III)-pyrophosphate concentration. Due to their prevalence in natural subsurface waters and their ability to rapidly oxidize TcO2, Mn(III)-ligand complexes pose a substantial liability to the environmental remediation of Tc-99.
Speaker #4
Ilana Szlamkowicz
University of Central Florida, Orlando, Florida
Comparative Study of Redox Transformations and Environmental Fate of Iodine as Influenced by Manganese Oxides at High and Low Concentrations
Abstract
Radioiodine, a by-product of nuclear fission, is a major component of the nuclear waste inventory. There are two common isotopes of this radionuclide, I-131 and I-129, with half lives of 8 days and 16 million years, respectively, with I-129 being the major concern due to its large half-life. Mishandling and poor management of radioactive waste in the past have resulted in the accidental release of radioiodine into the environment, with adverse effects in the quality of natural waters and the biosphere. In natural waters, iodine exists as iodide (I-), a highly mobile species that interacts very little with natural substrates, and iodate (IO3-), which is known to sorb, or even incorporate into minerals, leading to iodine natural attenuation.
The present study investigates the geochemical controls that high valence manganese oxides exert over the speciation, and consequently the fate, of iodide under environmentally relevant conditions. Manganese oxides are common natural minerals who have strong oxidizing capacity and this way they influence the fate and transport of redox sensitive risk driving contaminants. There have been few reports investigating the topic, but they studied iodine environmental fate and redox transformations under unrealistic conditions: parts per million levels of iodine initial concentration. In this study, we used birnessite (δ-MnO2), a common +3/+4 manganese oxide, and bixbyite (α-Mn2O3) as substrates in batch experiments containing iodide (I-) in high (10 mg L-1) and low, (20 μg L-1) initial concentrations. The concentration of iodine species for the experiments with high concentrations (ppm) were monitored using anion-exchange High Pressure Ion Chromatography (I- and IO3-) and UV-vis spectroscopy for I2, whereas for the experiments at ppb levels, the tandem Ion Chromatography-Inductively Coupled Plasma-Mass Spectrometry technique was used. Briefly, the sample is injected in the anion exchange column where it goes through chromatographic separation and then it is directed to the ICP-MS detector, which can provide detection levels of parts per trillion. This way speciation is performed through one analysis at levels that are environmentally relevant.
Experimental results revealed a completely different pattern for the redox transformations of iodide at ppm and ppb initial concentration levels. Specifically, at ppm concentrations there was a rapid and significant oxidation of I- to I2 and IO3- by the manganese oxides, whereas at ppb levels there was no oxidation of I-, despite the experiment having been performed under otherwise identical conditions. This experimental finding is very important because it demonstrates the disparity of iodine chemistry that is dependent on the concentration of iodine in the system. Specifically, at high concentrations the formation of I2 may lead to further reaction with organic matter and the formation of organo-iodine species, for example. Whereas, at environmental relevant conditions, this pathway is not anticipated. On the other hand, the absence of iodate (IO3-) eliminates a possible natural attenuation route. Other parameters tested were the effect of ionic strength, the presence of iron minerals, as well as other anionic co-contaminants.
Theme: Actinide Fate and Transport
Speaker #1
Hilary Emerson
PNNL
Characterization of PU and AM Mobility in Sediments from Beneath the Z-9 Trench at the Hanford Site, WA, USA
Co-authors: Carolyn I. Pearce, Kirk Cantrell, Michelle M.V. Snyder, Dallas D. Reilly, Edgar Buck, Jordan Zabrecky, and Vicky L. Freedman, PNNL; Calvin H. Delegard, Mavrik Zavarin, Annie Kersting, and Teresa Baumer, TradeWind Services
Abstract
Plutonium (Pu) and americium (Am) migration in the subsurface are profoundly influenced by redox conditions, pH, co-disposed and natural ions, complexing agents, and their combined effects on oxidation state and speciation. The objective of this work is to determine the mechanisms responsible for migration of Pu and Am in low pH, high-salt, organic-bearing waste streams released to the subsurface beneath the Z-9 Trench based on characterization of sediments following historical releases and controlled, laboratory-scale experiments with waste simulants.
Laboratory-scale experiments showed that historical Pu and Am (Eu as an analog for Am) mobility were relatively high. Kd partitioning coefficients for sorption in waste simulants ranged from 15 to 1530 mL/g for Pu and 0.04 to 43 mL/g for Eu, with the organic waste components decreasing mobility of both. However, characterization of sediments recovered from beneath the Z-9 Trench indicated that the current likelihood of release of Pu is low (desorption Kd range 0.2 x 106 to 18 x 106 mL/g) and is controlled by PuO2 or Pu phosphate [PuIIIPO4 or PuIV(HPO4)2] phases, depending on pH and available phosphate. Current release of Am from sediments was also low (desorption Kd range 3 x 106 to 9.9 x 109 mL/g). Its release appeared to be controlled primarily by AmIIIPO4, although release from neutral pH, deeper sediments may be controlled by adsorbed species or release of Am-241 in-grown on PuO2 from Pu-241. These data highlight that the mobility of Pu and Am in sediments impacted by acidic and organic wastes decreased significantly over time, potentially due to changes in Pu and Am speciation, secondary mineral precipitation, organic compound degradation, and aging processes.

Speaker #2
Teresa Baumer
Lawrence Livermore National Laboratory, Livermore, California
Pu Distribution in Simplified Waste Simulants and Minerals Relevant to the Hanford Site in WA, USA with Implications for Long-term Migration
Abstract
Beginning in 1943, approximately 1.85 x 1015 Bq (200 kg) of plutonium (Pu) were released into unlined cribs, trenches, and field tiles at the Hanford Site, Washington, USA. Between 1955 and 1962, over 4 x 106 liters of mixed Pu processing waste from the Plutonium Finishing Plant were pumped directly into the unlined 216-Z-9 trench. The waste contained two components (i) high salt (5 M NO3-, 0.6 M Al3+), acidic (pH 2.5) solutions and (ii) organic solvents: CCl4, TBP, DBBP, and lard oil. While most Pu was observed as precipitates within the first several centimeters, a small fraction of Pu was detected in the vadose zone at depths of up to 37 m. Pu in the subsurface has also been observed concurrently with TBP and in low pH sediments, which indicates that Pu may have traveled with its aqueous and organic co-contaminants as they moved through the vadose zone; yet, the mechanisms controlling long-term Pu mobility under these complex, dynamic conditions have yet to be determined.
In this study, Pu partitioning behavior was assessed between simulated organic, aqueous, and feldspar mineral phases using a series of binary and ternary batch experiments to develop a conceptual model for the transport of Pu in the subsurface. The experiments used either a deionized water or 5 M nitrate aqueous phase; a dodecane or 15% TBP in dodecane organic phase; and with or without the feldspar mineral albite. Partitioning behavior was assessed between pH 2-8 in all binary and ternary batch experiments. Significant concentrations of Pu only transferred from the aqueous phase into the organic phase when TBP was present (binary 5 M nitrate/15% TBP in dodecane and ternary 5 M nitrate/15% TBP in dodecane/albite) and occurred to a greater extent in high nitrate. Moreover, Pu partitioning into the organic phase was highly pH dependent with the highest Pu (~40%) partitioning at pH 2 and decreasing with increasing pH, likely due to the formation of Pu-TBP-nitrate complexes which are favorable at low pH. The major findings of this work show Pu at equilibrium with low pH, high nitrate waste and in the presence of a TBP/organic phase, will migrate in the organic phase as long as the low pH and high nitrate concentrations are maintained. Reducing nitrate concentrations or increasing pH will impact Pu mobility as less Pu will be present in the organic phase and may lead to Pu migration with the aqueous phase or sorption to Hanford sediments. This study helps further our understanding of Pu migration at the Hanford Site and other sites with similar waste releases.
Prepared by LLNL under Contract DE-AC52-07NA27344
Speaker #3
Naomi Wasserman
Lawrence Livermore National Laboratory, Livermore, California
Radionuclide Mobility in a Monomictic Pond
Abstract
Between 1961 through 1964, releases of radionuclides from Savannah River Plant facilities contaminated local streams and ponds which served as cooling reservoirs for R-Reactor. Pond B, an 0.87 km2 pond, received an estimated 33 MBq 238Pu, 430 MBq 239,240Pu, and an unknown amount of 137Cs (Pinder et al, 1992). Previous studies observed variations in Cs and Pu related to seasonal stratification of Pond B. From spring through early fall, thermally buoyant shallow waters remain oxic, while deeper, cooler waters become anoxic. The pond thermally overturns in late fall, resulting in a fully oxic water column until stratification is reestablished the following year. During the summer, Pu and Cs in filtered samples (<0.45 μm) are elevated in deeper anoxic water during reductive dissolution of Fe and organic rich sediments, while particle-associated Pu and Cs increase once the pond overturns (Alberts et al., 1986). Although this observation indicates organic particulate matter and Fe redox cycling can influence radionuclide mobility in Pond B, the mechanism(s) are not well-understood. In our study, we examine the impact of these biogeochemical processes on long-term transport of Pu and Cs in the Pond B system.
We conducted two sampling campaigns in June 2019 and March 2020 within a transect from the inlet to the outlet of Pond B. Water samples were collected at five locations at 1 m depth increments. Samples were analyzed for physicochemical water parameters, concentrations of cations, anions, Fe(II), TOC, 137Cs, Pu, 240Pu/239Pu and microbial community. During stratification, dissolved oxygen levels decreased with depth while Fe(II), TOC, Pu, and Cs increased. We hypothesize that reductive dissolution of Fe-(oxy)hydroxide phases in the sediments, remobilization of organic matter associated with Pu and Fe, and upward diffusion of Fe, Cs, and Pu to the thermocline lead to this elevation. In the thermocline, microbes likely influence Fe-(oxy)hydroxide formation and adsorbed or co-precipitated radionuclides. Notably, we observed that putative Fe(II) oxidizers subsisted throughout the water column, with a select few preferring certain depths, indicating the likely formation of biotic Fe-(oxy)hydroxides. We also observed an increase in relative abundance of putative Fe(III)-reducing bacteria below the thermocline, suggesting microbe-mediated mineralization of Fe-(oxy)hydroxides. This seasonal remobilization and scavenging results in thorough cycling of Pu in Pond B, as all water samples are isotopically identical to upstream sediments. Future work will focus on the potential migration of particulates associated with radionuclides from Pond B to downstream sediments.
Prepared by LLNL under Contract DE-AC52-07NA27344.
Speaker #4
Mariah Doughman
Florida International University, Miami, Florida
Impact of Major Groundwater Components on the Adsorption of Uranium (VI) to Hanford Formation Sediment
Co-authors: Yelena Katsenovich, Leonel Lagos, and Kevin O’Shea, Florida International University; Hilary Emerson, James Szecsody, and Nik Qafoku, PNNL
Abstract
Hexavalent uranium [U(VI)] mobility in soils and sediments is heavily influenced by its aqueous speciation as this determines the potential for adsorption, precipitation/incorporation, and redox reactions. Previous studies have found that the mobility of U(VI) is increased in the presence of calcium and carbonate aqueous species, due to the formation of U(VI) neutral and negatively charged species. The objectives of this research effort are to provide (i) a better understanding of the species-dependent mechanisms of U interaction with sediments; (ii) the necessary parameters to predict U mobility in the vadose zone; and (iii) the technical basis for Monitored Natural Attenuation at the Hanford Site. Hanford formation sediments were used to study the extent and rate of U(VI) adsorption at relevant aqueous concentrations (50-9000 µg/L), in the presence of major groundwater components (NaHCO3, KHCO3, MgSO4•7H2O, MgCl2•6H2O, CaCl2•2H2O, pH: 7.90 ± 0.03), and as a function of time and concentration. The results from geochemical modeling indicate the dominant presence of neutral Ca uranyl carbonate species in the aqueous phase. The results from a series of batch experiments conducted at room temperature and in the presence of air indicated low U(VI) adsorption (Qmax=5.6 mg/g). Removal percentage and log Kd values at equilibrium were also minimal ranging from 48% and 0.21 L/Kg to 71% and 0.43 L/Kg over the entire concentration range. These results illustrate that complexation of U(VI) with Ca and carbonate ions negatively affected the extent of adsorption, increasing its mobility under site relevant conditions at the Hanford Site.

Speaker #5
Jose Cerrato
University of New Mexico, Albuquerque, New Mexico
Transport and Remediation of Metal Mixtures in Mine Wastes
Co-authors: Carmen Velasco, Sumant Avasarala, Eliane El Hayek, Mehdi Ali, and Adrian Brearley, University of New Mexico; Juan Lezma-Pacheco, Stanford University
Abstract
The transport and potential remediation approaches for metal mixtures in mine wastes from sites located in tribal land in the Southwestern US was investigated by integrating laboratory experiments, microscopy, and spectroscopy. Metal release from these mine wastes could pose potential health risks for neighboring communities. Spectroscopy analyses suggest that U-V and U-organic-rich phases are present in abandoned mine wastes; the dissolution of these phases is relevant to U, As, and V transport. Remediation approaches using naturally occurring minerals is currently being researched for the immobilization of these metal mixtures. Additionally, calcium in carbonate water at circumneutral pH facilitates the transport of U(VI) in plant roots which could be useful for metal uptake. These results are relevant for uranium transport and remediation in the proximity of mine wastes and mineralized deposits.
Theme: Remediation
Speaker #1
Pietro Paolo Falciglia
University di Catania, Catania, Sicily
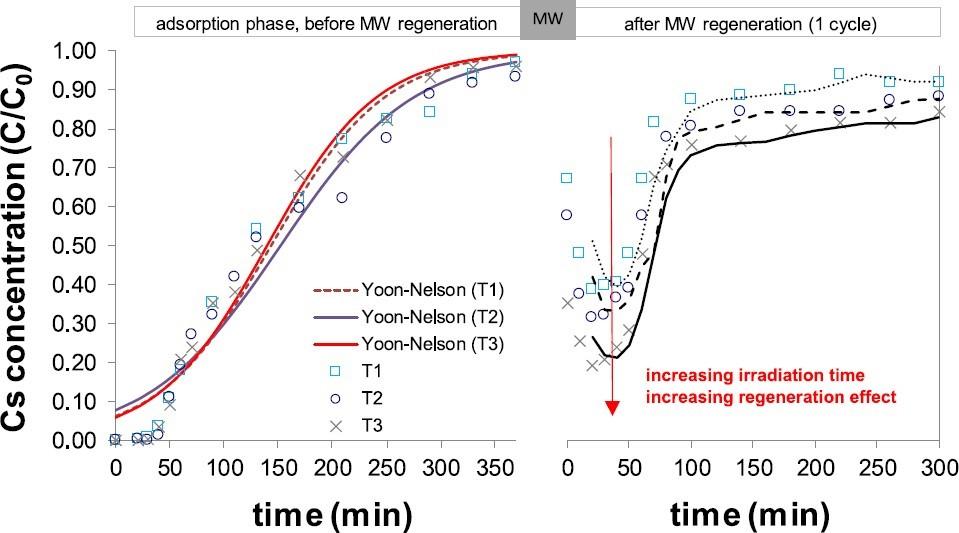
Microwave-based Regenerating Permeable Reactive Barrier (MW-PRB) For CS Removal from Groundwater
Co-authors: Erica Gagliano, LNS – INFN; Vincenza Brancato, CNR - ITAE – Istituto di Tecnologie Avanzate per l’Energia; Guglielmo Finocchiaro, Alfio Catalfod, Guido De Guidi, Stefano Romano, Paolo Roccaro, and Federico G. A. Vagliasindi, University di Catania
Abstract
Reduced longevity of Permeable Reactive Barriers (PRBs) due to filling material saturation is the major problem affecting full-scale treatments (Ghaeminia and Mokhtarani, 2018). As a consequence, a frontier research investigation is coupling PRBs with regeneration processes in order to increase their longevity (Huang et al., 2017). The present study evaluates the concept of a novel microwave based regenerating permeable reactive barrier (MW-PRB) system as in situ- regenerating technology with focus on Cs-contaminated groundwater.
Granular activated carbon (GAC) was selected as adsorptive materials to conduct batch and column MW-regeneration experiments. Column tests were carried using a custom-made setup (GAC-filled column implanted in a MW oven cavity) simulating the in situ regeneration process at different irradiation times. Data were elaborated for technical and economic considerations.
Batch tests results revealed a very rapid increase in GAC temperature during MW irradiation up to ~680 °C. Physical characterization after multiple regeneration cycles revealed the preservation of GAC life span and regeneration efficiency during MW irradiation. Results from column tests confirms that GAC can be regenerated by MW also in dynamic condition. The highest Cs removal of ~80% was observed when the regeneration time of 15 min was applied.
Speaker #2
Callum Robinson
University of Manchester, Manchester, U.K.
Approaches to Sellafield Groundwater Radionuclide Remediation - In Situ Phosphate Mineralisation
Co-authors: Sam Shaw, Jonathan R. Lloyd, and Katherine Morris, University of Manchester; James Graham, National Nuclear Laboratory
Abstract
Historical activities at Sellafield, the UK’s largest legacy nuclear facility, have led to leakages of radionuclides into the site sub-surface causing radioactively contaminated land. Of these leaked radionuclides, strontium-90 (90Sr) is one of the more challenging from a remediation perspective due to its relatively high mobility in the Sellafield subsurface. The development of remediation strategies for 90Sr are therefore important in maintaining the stewardship of the site in the medium to long term. Calcium phosphate containing minerals such as hydroxyapatite (HAp) have been shown to incorporate some radionuclides within their structure. In previous studies, conducted at the Hanford nuclear site, USA, aqueous injection of HAp-generating solutions have been shown to reduce the amount of mobile 90Sr within contaminated sediments on both a laboratory and field scale.
In this study, three key phosphate amendment techniques (calcium citrate / sodium phosphate, glycerol phosphate and polyphosphates) were tested using microcosm experiments with the aim of developing a targeted remediation toolkit for 90Sr remediation at Sellafield. Aqueous geochemical results suggest all three amendment solutions removed Sr from solution at an enhanced rate when compared to the sediment sorption control, with the calcium citrate / sodium phosphate technique removing 92% of the initial concentration of Sr, glycerol phosphate 65% and polyphosphate 54%. Scanning electron microscopy in conjunction with EDX spectroscopy conducted on sediments after treatment with the phosphate amendment techniques showed the presence of strontium containing calcium phosphate phases deposited on the surface of larger sediment grains. XAS conducted on the treated sediments shows evidence of a Sr-P shell suggesting that Sr incorporation into the calcium phosphate phases has occurred. These data will be discussed in the context of providing a toolkit for remediation at Sellafield.
Speaker #3
Amanda Lawter
PNNL
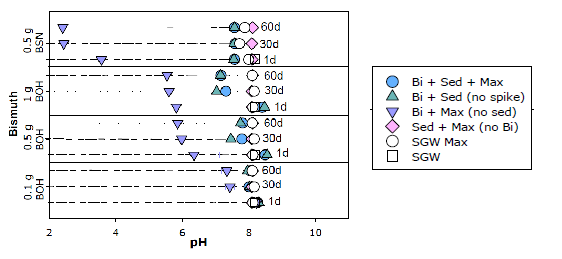
Sediment Interactions with Layered Bismuth Materials and Implications for Subsurface Contaminant Remediation
Co-authors: Nikolla P. Qafoku, Tatiana Levitskaia, and Carolyn I. Pearce, PNNL
Abstract
In this study, two bismuth (Bi) materials, Bi oxyhydroxide (BOH) and Bi subnitrate (BSN), were evaluated for their ability to immobilize contaminants of concern, technetium (Tc-99), uranium (U), radioiodine (I), and hexavalent chromium (CrVI), in the presence of sediments and under geochemical conditions relevant to the Hanford nuclear site, located in Washington State, USA. The effect of Bi-based material-to-sediment ratio was investigated to determine the quantity of Bi-based material required to achieve removal of the contaminants in the presence of sediment. Two concentrations of contaminant combinations were investigated, representing distributions of Hanford subsurface contaminants. To characterize the impact of sediments Bi-based material performance, changes in solution chemistry were measured and solid phases characterized.
In experiments without sediment, aqueous pH decreased to 2.4 with BSN or 5.9 with BOH, but, in the presence of sediment, the pH was buffered at 7-8 (Figure 1). A solution-to-Bi-based material ratio of 1000 readily removed Cr(VI) and U from the aqueous phase at concentrations of 0.05 and 2.3 mg/L, respectively. More Bi-based material (i.e., a ratio of 100-200) was required for complete removal of Tc-99 and I; changes in solution chemistry suggest that this is due to competition with other ions, including chloride and sulfate. The presence of sediment decreased removal of Tc-99 and I by BOH by 91% and 21%, and 33% and 93% for BSN, respectively. The presence of sediment increased removal of U, with 100% U removed in the BSN and sediment test, and only 17% of U removed in the absence of sediment, likely due to the changes in U speciation at lower pH; U removal in BOH tests was near 100% with or without sediment. These experiments demonstrated that Bi-based materials have the potential to rapidly remove co-located contaminants over a range of geochemical conditions, including the presence of subsurface sediments.
Speaker #4
Ajay Karakoti
The University of Newcastle, New Castle, New South Wales, Australia
Moderation of Nanoparticle Toxicity by Abiotic Factors
Co-authors: Krupa Kansara and Ashutosh Kumar, Ahmedabad University
Abstract
Technology has always been ahead of toxicology, as the environmental and health issues take the centre stage only after the technology/products are released into the market. It is thus unsurprising that the nanotoxicology only caught up to nanotechnology several years later. The extensive use of Engineered Nanomaterials (ENMs) in several consumer products has resulted in their release in the aquatic environment. The build-up of ENMs has the potential to threaten the natural ecosystem especially the aquatic ecosystem. ENMs entering the aquatic ecosystem undergo a dynamic transformation as they interact with organic and inorganic constituents present in the aquatic environment. Recent studies on the ecotoxicity of ENMs have shown that the ENMs can undergo transformation in aquatic system by interacting with abiotic (such as, natural organic matter (NOM) and clay minerals) and biotic factors and attain an environmental identity. These studies have focused on the influence of abiotic factors on the bioavailability and subsequent ecotoxicity of ENMs to aquatic organisms though usually in presence of one of the major abiotic factors. This talk will focus on our efforts on comparing the interaction of two abiotic factors (humic acid and montmorillonite clay) on the two most popular NPs (TiO2 NPs and CuO NPs) to understand their impact on the biological and ecological fate and toxicity. The physiochemical properties were characterized via stability, pH, agglomeration and DLVO studies in the relevant media. The ecotoxic response of ENMs in presence and absence of abiotic factors was compared by understanding their effect on the development of zebrafish fish embryos. It was found that even though, CuO NPs were more stable towards agglomeration than TiO2 NPs in presence of abiotic factors at most pH values however, the toxic potential of CuO NPs was 4 to 5 fold higher in zebrafish embryos as compared to TiO2 NPs which was directly related to the stability of NPs. The presence of HA and clay interfered with the interaction of both CuO and TiO2 NPs with zebrafish embryos and both NPs showed least toxicity in co-presence of combination of HA and clay. Even though a complete mechanism is under investigation however, it can be proposed that humic acid and clay minerals have the potential to reduce the toxic effects exerted by ENMs on developing zebrafish embryos irrespective of the ENM chemistry.
Speaker #5
Muthu Prabhu Subbaiah
Hanyang University, Seoul, Republic of Korea
Synthesis of Carboxylate-rich Graphene Oxide Decorated Magnetic Spinel Materials for Effective Adsorption of Perfluorooctanoic Acid from Water
Co-authors: Alam Venugopal Narandra Kumar, S.SD. Elanchezhiyan, and Chang Min Park, Kyungpook National University; Byong-Hun Jeon, Hanyang University
Abstract
Perfluorooctanoic acid (PFOA) is a persistent organic contaminant, and it has recently been recognized as a contaminant in aquatic environments worldwide because of its effects on human health. Herein, we report the synthesis of graphene oxide decorated with highly stable lanthanum-substituted manganese ferrites (LMF) at various concentrations (LMFx%@GO).
After the substitution of lanthanum on MF nanorods, the surface charge density and the crystalline nature were altered significantly, as supported by the zeta potential, transmission electron and scanning electron microscopy images, and powder X-ray diffraction patterns. Among the concentrations of X = 25, 50, and 75% wt./wt., LMF75%@GO exhibited the highest PFOA removal densities. This substantially enhanced the adsorption densities of the nanohybrids for PFOA from water. Electrostatic interaction and hydrogen bonding played vital roles in the adsorption of PFOA, as evidenced by various spectro-analytical techniques, including Fourier transform infrared, X-ray photoelectron spectroscopy, and transmission electron microscopy energy-dispersive X-ray spectroscopy. Several conditions such as contact time, pH, initial concentration, and quantities of LMF nanoparticles were evaluated to provide information about the PFOA removal process. The spent adsorbents were separated easily by applying an external magnetic field. The results of this study provide useful insights into PFOA removal from real water systems.