Technology Overview
Conventional metabolomics and small molecule identification approaches have demonstrated immense value for disease diagnosis, evaluation of environmental exposures, and discovery of novel molecules, but traditional approaches fail to identify small molecules in biological and environmental samples. Such approaches either seek to identify specific compounds or seek to identify as many compounds as possible, relying on authentic, pure reference materials (standards) for unambiguous chemical identification. These approaches are, therefore, limited to the subset of molecules for which these standards exist. For example, less than two percent of compounds found in typical exposure chemical databases are available commercially in pure form as a standard. Current approaches also struggle to match the speed of the high-performance computing needed to accurately quantify a sample.
Researchers at Pacific Northwest National Laboratory are advancing standards-free metabolomics—the identification of small molecules without reliance on standards—using calculated chemical properties and associated matching with multiple experimental attributes. PNNL’s unique approach relies on multiple experimental data types, including accurate mass, isotopic distribution, collisional cross section (a structural property derived from ion mobility measurements), mass fragmentation patterns, data from multiple adducts, and nuclear magnetic resonance chemical shifts. These values are then compared to entries in in silico libraries, leveraging experimental, instrumental, and computational innovations.
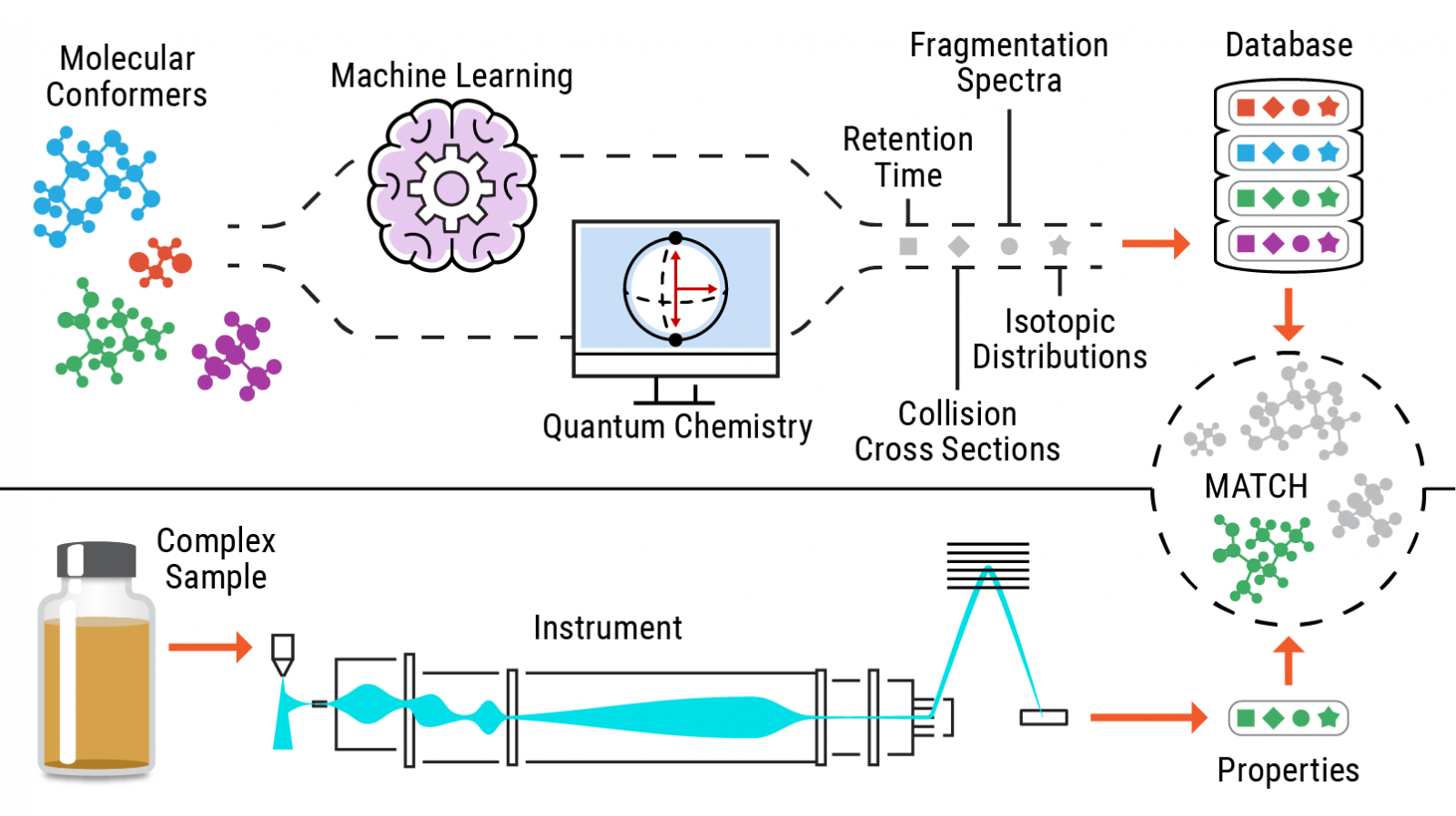
An illustration of the standards-free identification process of metabolites.
The approach includes four key tools
- Data Extraction for Integrated Multidimensional Spectrometry (DEIMoS), a modular software tool that can extract features from data collected on multi-dimensional analytical platforms.
- The in silico chemical library engine (ISiCLE), a high-performance-computing-friendly approach for generating predicted chemical properties.
- The Multi Attribute Matching Example (MAME), which matches properties based on various chemical attributes.
- DarkChem, a variational autoencoder that learns a continuous numerical or latent representation of molecular structure, which can characterize and expand reference libraries.
The tools involve state-of-the-art first-principles simulation, distinguished by use of molecular dynamics, quantum chemistry, and ion mobility calculations, to generate predictions of chemicals that may be in the sample. In the case of ISiCLE, scientists optimized the popular MOBCAL code to predict collision cross sections, improving computational efficiency by more than two orders of magnitude. Using DarkChem, a trained deep learning network can predict chemical properties directly from structure, as well as generate candidate structures with chemical properties similar to an input.
Together, the tools represent a promising method to enable identification of molecules in biological and environmental samples and offer a high-performance-computing-friendly alternative to standards-based chemical identification. The tools were valuated through the U.S. Environmental Protection Agency’s Non-Targeted Analysis Collaborative Trial (ENTACT). The inter-laboratory challenge provided a consistent set of verified, blinded synthetic mixtures for the objective testing of non-targeted analytical chemistry methods. The evaluation demonstrated the potential of PNNL’s standards-free small molecule identification tools, particularly the value of using calculated, orthogonal properties and multi-attribute matching to increase confidence in compound identification. The approaches significantly reduce reliance on standards and open the door to the identification of up to 90 percent more chemicals in samples.
APPLICABILITY
PNNL’s tools can be used to improve the identification of metabolites and other chemicals in biological and environmental samples. They have been successfully used to identify environmental degradation products, separate molecular isomers, and decode complex blinded mixtures of exposure chemicals. They can also be used to enable prediction of nuclear magnetic resonance chemical shifts and spin-spin couplings. In addition, the tools can be applied to novel molecular design to refine for a particular function or chemical feature. For example, recent research was able to narrow millions of possible molecules down to about 20 first-priority candidate molecules. Open-source versions are available on Github.
Advantages
- Highly accurate first-principles approaches for predicting chemical properties without reference standards
- High throughput and comprehensive
- Accurate identification of metabolites and other chemicals in biological and environmental samples