
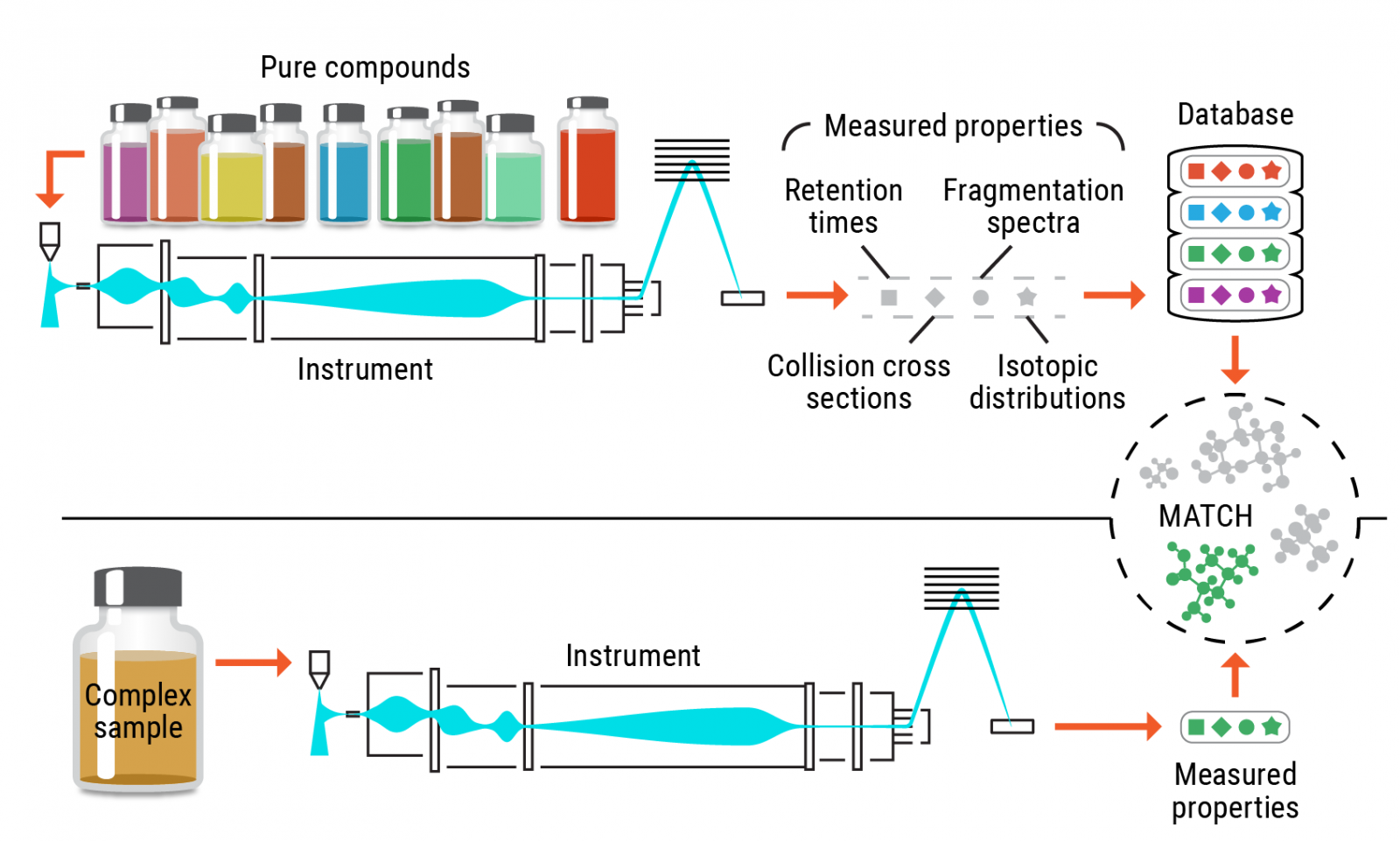
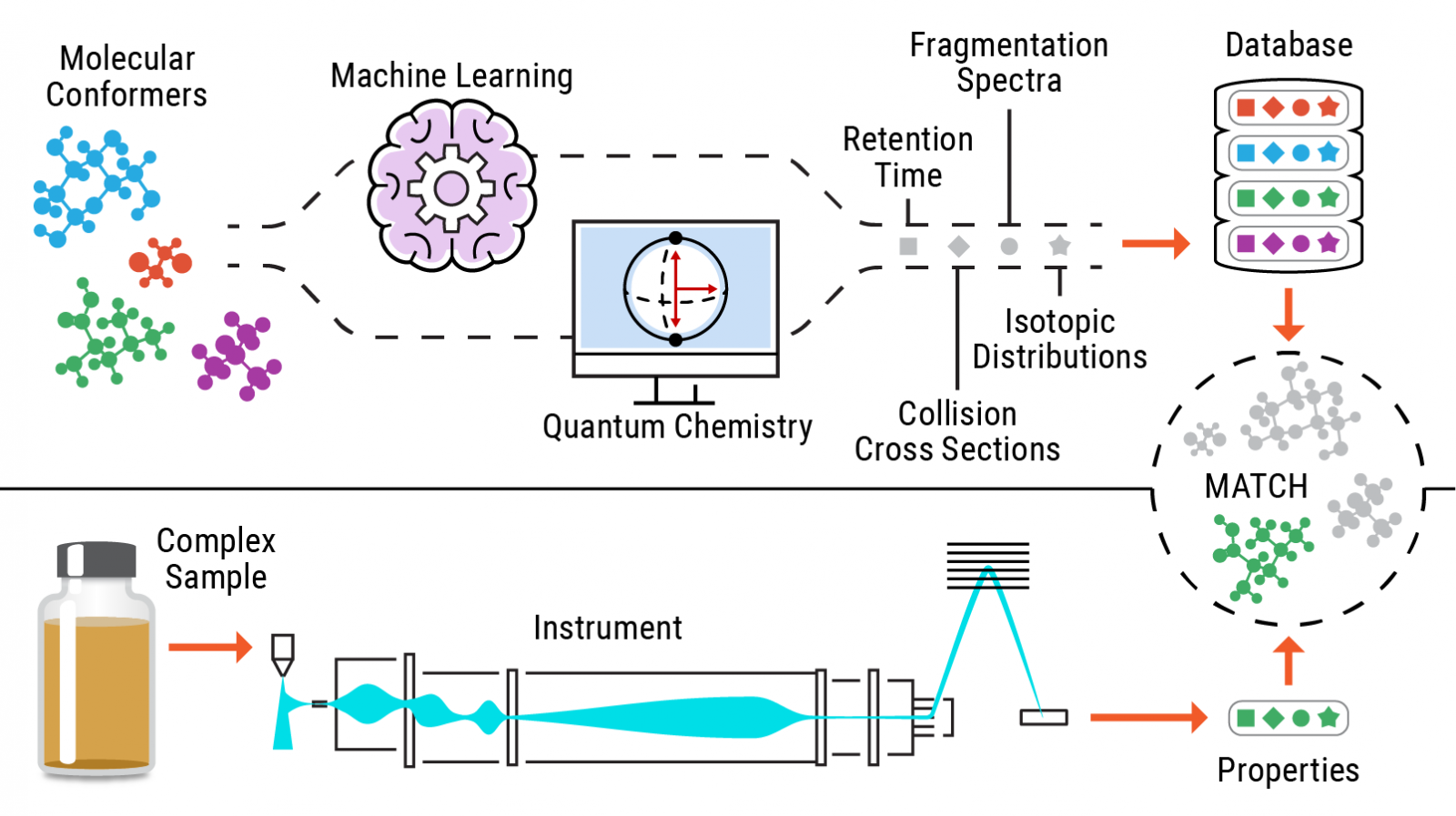
###
About PNNL
Pacific Northwest National Laboratory draws on its distinguishing strengths in chemistry, Earth sciences, biology and data science to advance scientific knowledge and address challenges in energy resiliency and national security. Founded in 1965, PNNL is operated by Battelle and supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit the DOE Office of Science website. For more information on PNNL, visit PNNL's News Center. Follow us on Twitter, Facebook, LinkedIn and Instagram.