Return to Data and Integration Activities
PI: Jenna Pope
Project Team: Michael Ashby, Sutanay Choudhury
Project Collaborators: Peter Sushko, Malachi Schram
Project Term: 2 years (fiscal year 2021 and fiscal year 2022)
Key Science Questions:
How can we use deep learning to predict the dynamics of chemical and material systems?
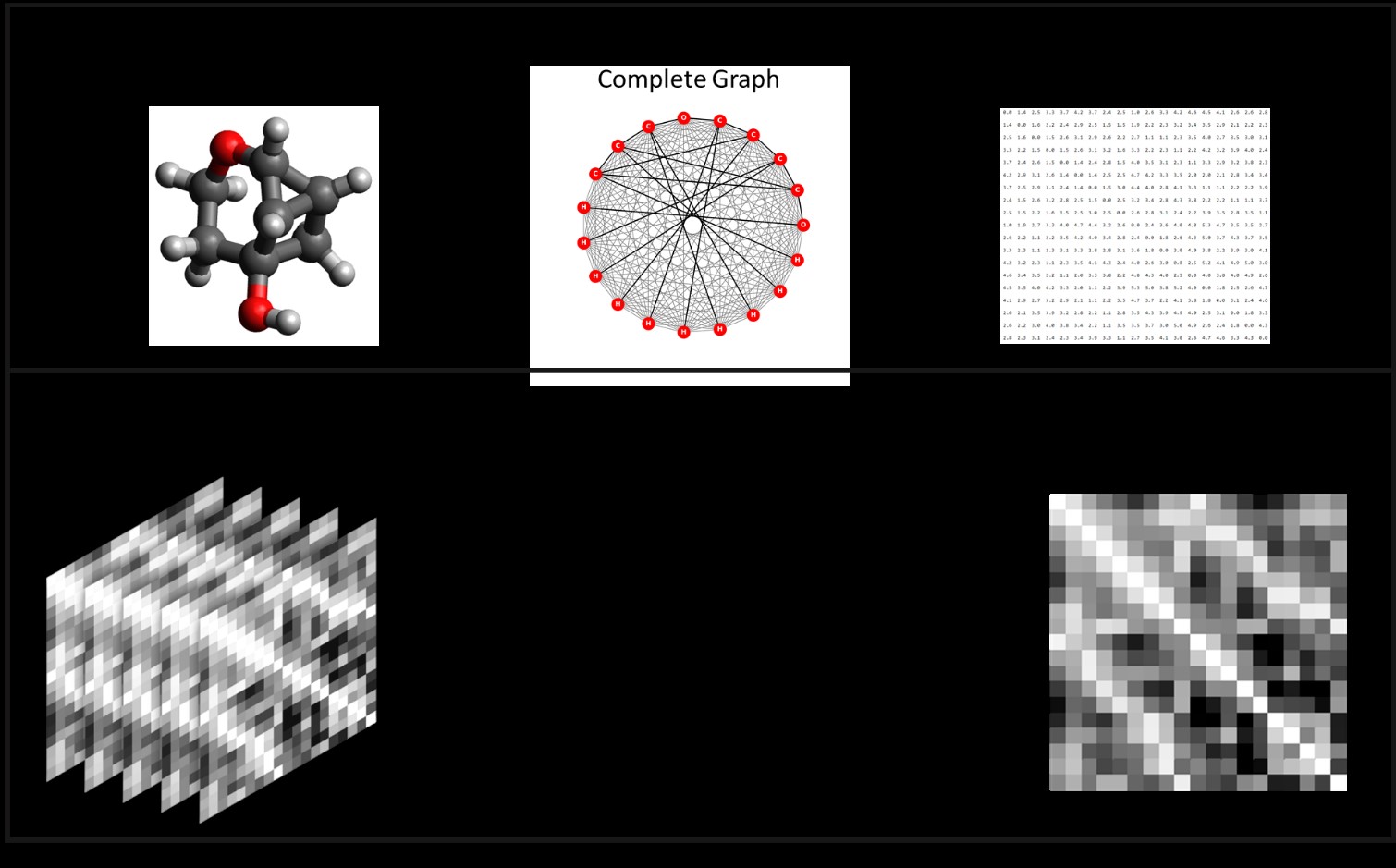
How can we further the use of graph neural networks for chemical systems by taking into account nonbonded atom-to-atom distances in addition to bond distances?
Can we predict system energy with high-accuracy using neural networks from our predated graphs?
Project Description:
What need does your project address?
Simulations of large material systems are often unfeasible due to limited computational resources. Deep learning can provide predictions of material dynamics more quickly, opening the potential for modeling large systems.
How is your approach different than what is currently being done?
Most approaches to predict chemical and material dynamics using deep learning focus on the development of machine-learning potentials for molecular dynamics (MD) simulations, predicting the energy and forces from a single structure and relying on MD hardware to push the dynamics forward. Our method instead takes a cue from dynamic graph networks and predicts the structural and energetic change given a short sequence of prior structures. In the grand scheme, this would allow a researcher to perform a short simulation through traditional methods and then extrapolate to longer time scales using deep learning.
How will you gauge your project’s success?
We are developing the code using the ISO17 dataset, which consists of MD simulations of C7O2H10 isomers. Success with this set will be prediction of atom-to-atom distances within 0.1 Å and energy within 1 eV over a 100 fs time scale. We expect to publish a paper on this architecture.
We will then move to materials systems relevant to the Chemical Dynamics Initiative.
First, we will examine geometry relaxation of palladium atoms on a transition metal dichalcogenide surface. We will aim to end the dynamics on the correct structure and predict the energy within the same error tolerance. We will publish a follow up to the prior architecture paper detailing its implementation for materials systems and geometry relaxation, as compared to MD of small molecules.
We will also explore adding physics information into the graph network, as we suspect this will increase the accuracy and/or allow training on a decreased amount of data. We will focus on the material dataset generated by Malachi’s project. Success will look like an updated model that accounts for physics and has the same or better error tolerance as described above. We expect to collaborate on a publication with the improved physics-informed network.