Single-Crystal to Single-Crystal Insertions of Carbon Dioxide Into Bridging Copper Hydride Bonds
Studying single crystal reactions provides molecular-level insight into catalytic processes
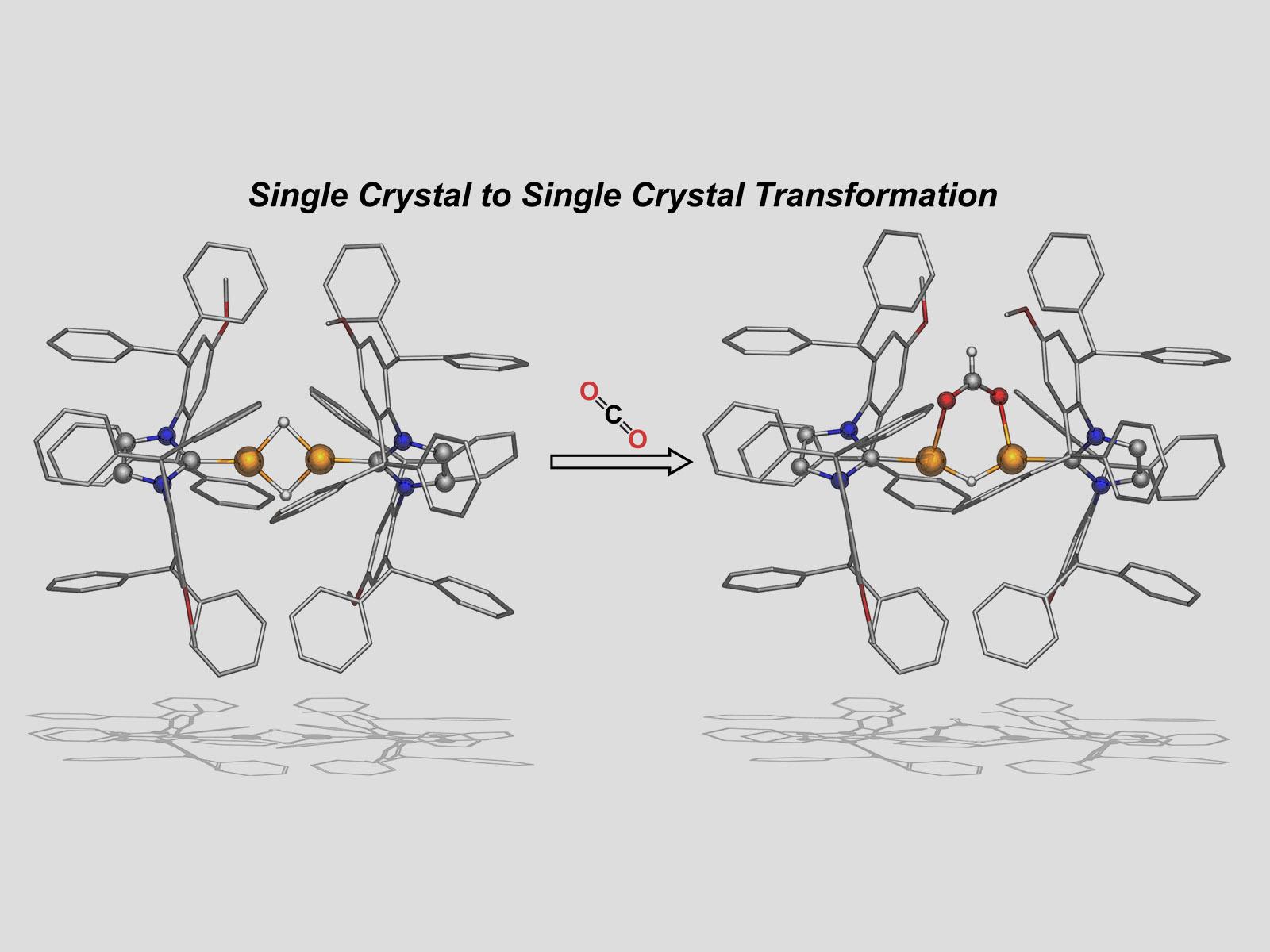
Carbon dioxide inserts into copper–hydrogen–copper bonds in a solid crystal.
(Image by Ba Tran | Pacific Northwest National Laboratory)
Published: September 8, 2023
Patrick E., M. Bowden, J. Erickson, R. M. Bullock, and B. Tran. 2023. Angew. Chem. Int. Ed. [DOI: 10.1002/anie.202304648]