MixPI: A Path-Integral Software for Large Systems
New software enables the study of nuclear quantum effects in large, condensed phase systems up to 10× faster than traditional methods
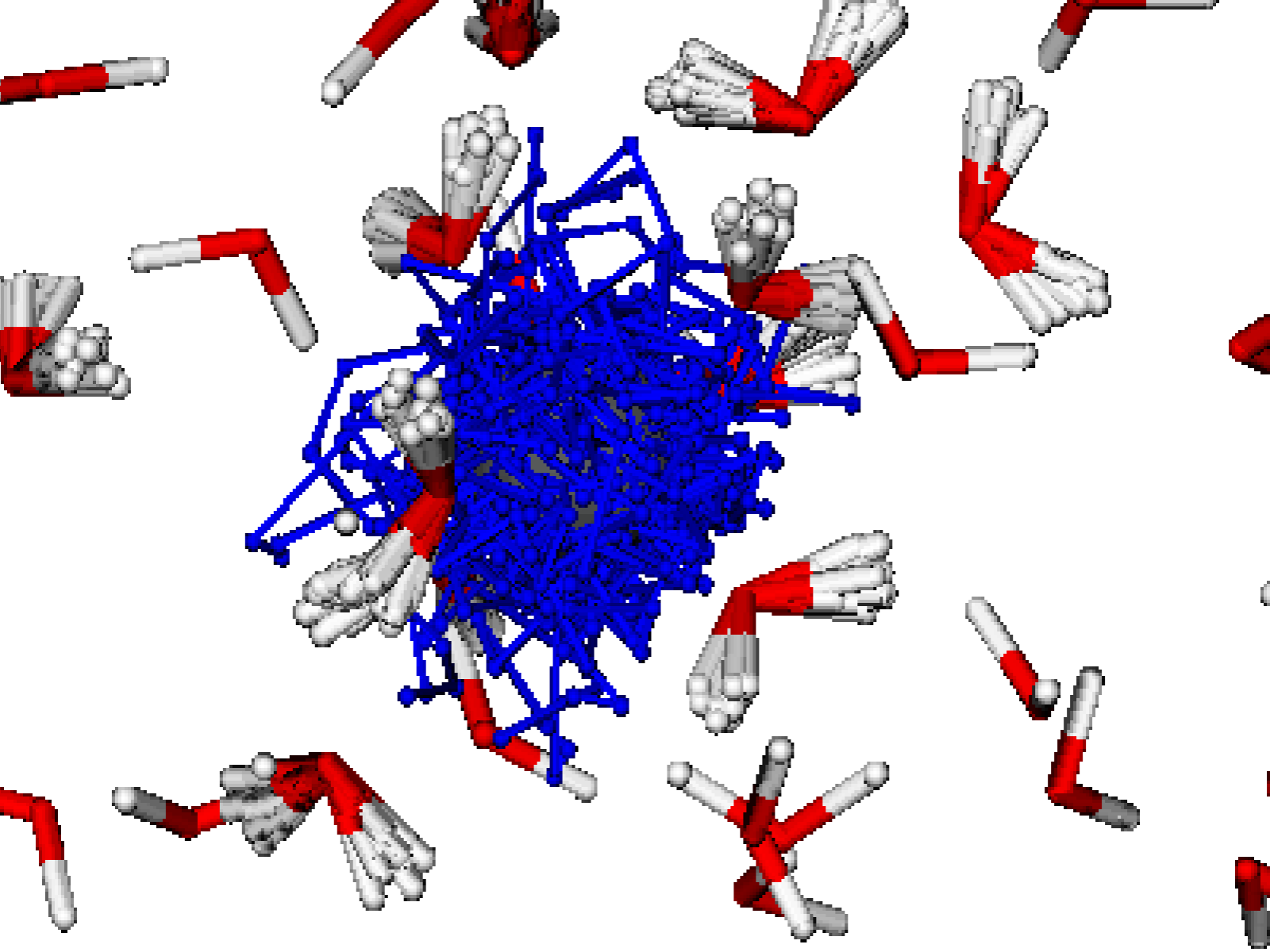
A solvated metal ion M3+ (gray) with an explicit ring polymer electron (blue) solvated by a first solvated shell of ring polymer water (red and white) and remaining waters treated classically. MixPI can use different sized ring polymers to represent different atoms in a simulation, significantly reducing the computational expense of path-integral calculations. The width and size of the ring polymers show the relative strength of the nuclear quantum effects for that atom.
(Image by Britta Johnson | Pacific Northwest National Laboratory)
Published: June 30, 2026
Johnson, B. A., S. Bu, C. J. Mundy, N. Ananth. 2026. “MixPI: Mixed-time slicing path integral software for quantized molecular dynamics simulations,” J. Chem. Phys. 164 (21), 212501. DOI: 10.1063/5.0327460.